
Our research group employs a variety of theoretical and computational approaches to investigate the electronic, thermal, and magnetic properties of solids, with a particular emphasis on strongly correlated materials.
We primarily focus on the theoretical study of systems in which electrons exhibit strong electronic correlations. Understanding the electronic properties of these systems requires approaches that go beyond the single-particle approximation commonly used in other areas. Using advanced many-body techniques such as DFT+DMFT and GW+DMFT, we study the electronic structure of transition-metal oxides and other quantum materials. Our goal is to uncover the underlying physical mechanisms responsible for emergent phenomena such as metal–insulator transitions, unconventional superconductivity, orbital differentiation, and correlated metallic phases.
We have also investigated the thermal properties of complex oxides by explicitly treating phonon–phonon interactions and anharmonic effects, employing the mode-coupling formulation of heat transport. In addition, we are engaged in the development of computational tools for DFT+DMFT calculations and for simulating x-ray absorption spectroscopy (XAS) in complex oxides and heavy-fermion systems.
Research topics include the electronic and thermal properties of complex oxides, the electronic behavior of unconventional superconductors and intermetallic compounds, and materials with potential topological phases.
Research topics:
- Strongly correlated materials
- Metal–insulator transitions and other emergent phenomena in strongly correlated materials
- Computational modeling of materials using DFT, GW, DFT+DMFT, and GW+DMFT methods
- Electronic structure and spectroscopic properties of strongly correlated materials, including transition-metal oxides, intermetallic compounds, and quantum materials
- Electronic correlation effects in solids
- Thermal properties of anharmonic solids
- X-ray absorption spectroscopy (XAS) modeling
- Development of computational tools
Research Highlights:
Anomalous Glassy Thermal Conductivity in BaBiO3:
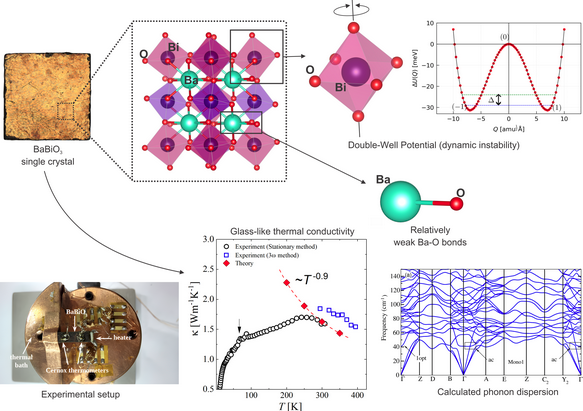
In collaboration with experimental researchers from the Institute of Physics at USP, we investigated the thermal properties of BaBiO₃ , a semiconducting, non-magnetic perovskite oxide and the parent compound of the high-Tc bismuthate-based cubic superconductors Ba₁₋ₓKₓBiO₃. Known since the 1970s, BaBiO₃ has recently regained attention as a promising candidate for engineering novel functional heterostructures, driven by theoretical predictions that electron-doping could induce a topological insulating state. Remarkably, despite its well-ordered crystalline structure, BaBiO₃ conducts heat as poorly as glass, an extremely rare behavior among solid materials.
For further information:
“Anomalous Glassy Thermal Conductivity in a Perovskite Bismuthate Induced by Structural Dynamic Instability”, Alexandre Henriques, Mariana S. L. Lima, Gøran J. Nilsen, Matthias J. Gutmann, Steffen Wirth, Walber H. Brito, Valentina Martelli, Advanced Science, e02379 (2025).
Interplay between electronic correlations and topology in iron based supercondutors (FeSe1−𝑥Te𝑥):
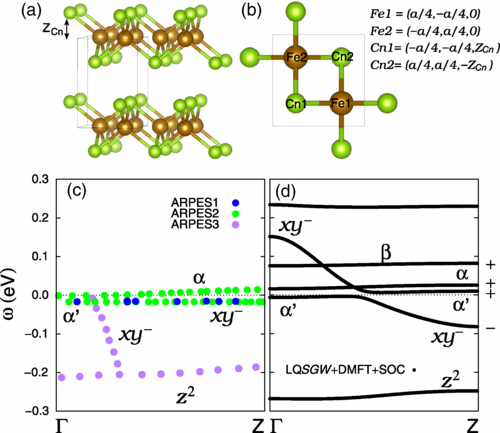
Our research on the iron-based superconductor FeSe₁₋ₓTeₓ explores its remarkable role as a platform for two frontier phenomena in condensed matter physics: the emergence of Majorana zero modes, which are promising candidates for topologically protected quantum bits, and the realization of an orbital-selective Mott transition. By employing advanced first-principles methods, specifically, linearized quasiparticle self-consistent GW combined with dynamical mean-field theory, we uncovered that the material’s topologically protected Dirac surface state has a strong Fe(dₓᵧ) orbital character. Interestingly, the proximity to the orbital-selective Mott transition plays a dual role: it promotes the emergence of the topological surface state by shifting the Dirac cone near the chemical potential, but also suppresses Z₂ topological superconductivity when the system gets too close to the Mott phase. Our results not only shed light on the delicate balance between strong electronic correlations and nontrivial topology in FeSe₁₋ₓTeₓ, but also provide a framework for investigating similar effects in other iron-based superconductors.
For further information:
“Orbital-Selective Mott Transition Effects and Nontrivial Topology of Iron Chalcogenide”, Minjae Kim, Sangkook Choi, Walber Hugo Brito, Gabriel Kotliar, Phys. Rev. Lett. 132, 136504 (2024).
Correlated Oxide superlattices (La₂/₃Sr₁/₃MnO₃/YBa₂Cu₃O₇):
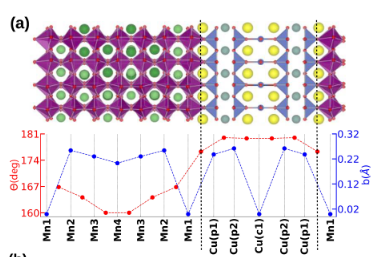
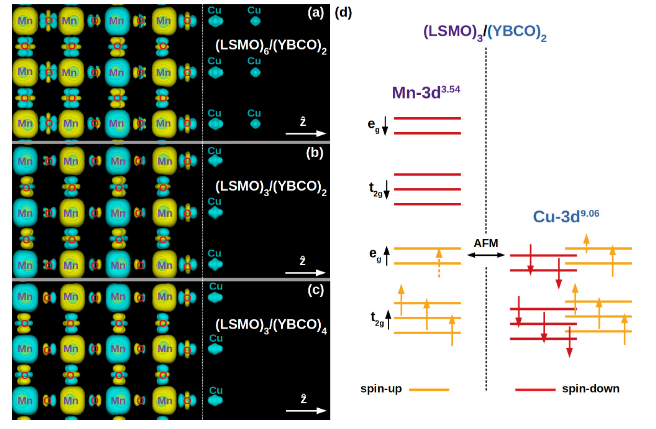
We investigate the interplay between strain, thickness, and interfacial effects in La₂/₃Sr₁/₃MnO₃/YBa₂Cu₃O₇ (LSMO/YBCO) superlattices from a first-principles perspective. Our results reveal that epitaxial strain and the presence of LSMO reduce the buckling of the interfacial CuO₂ planes and drive electron transfer from LSMO to YBCO, with the Cu 3d valence showing only a slight dependence on LSMO layer thickness. Remarkably, we find that the CuO₂ planes near the interface develop an in-plane ferromagnetic ground state due to local moments on copper atoms. These moments are not directly linked to charge transfer but instead arise from Mn 3d–O 2p–Cu 3d hybridization, which is confined to the interface region. This induced interfacial magnetism could play a significant role in controlling the superconducting state of LSMO/YBCO superlattices, opening possibilities for engineering oxide heterostructures with tunable functionalities.
For further information:
“Interface and thickness effects in La2/3Sr1/3MnO3/YBa2Cu3O7 superlattices”, V. A. M. Lima, M. C. O. Aguiar, N. C. Plumb, M. Radovic, W. H. Brito, Phys. Rev. B 109, 045128 (2024).
Effects of hole doping and electronic correlations in iron based supercondutors (BaFe2As2):
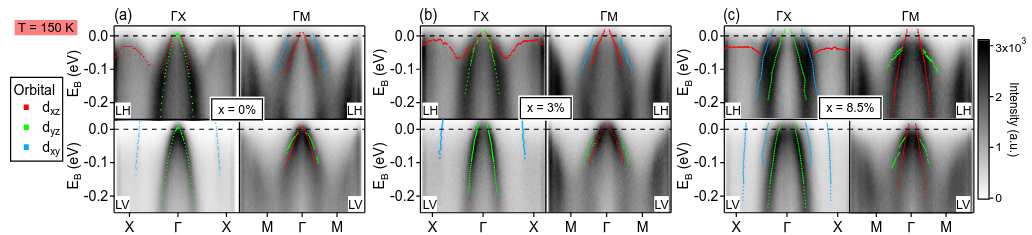
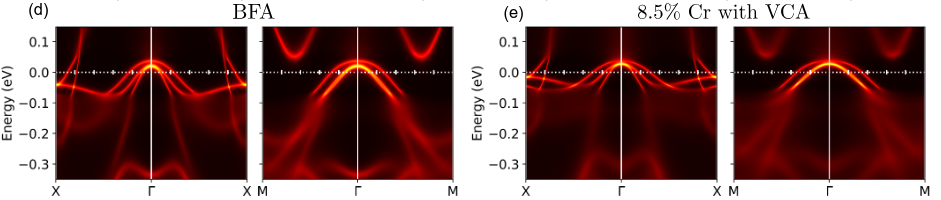
We have conducted a comprehensive study of the electronic properties of Cr- and Mn-substituted BaFe₂As₂ (CrBFA and MnBFA), two iron-based compounds where superconductivity is notably absent across a wide range of compositions. Using angle-resolved photoemission spectroscopy (ARPES) combined with density functional theory plus dynamical mean-field theory (DFT+DMFT), we found that Cr substitution effectively hole-dopes the states near the Fermi surface, a behavior well captured by the virtual crystal approximation, while also displaying clear signatures of Hund’s correlations in bands with dominant dyz orbital character. In MnBFA, Mn substitution causes only a slight and orbital-selective reduction of the electron pockets but strongly increases the electronic scattering rate, driving the system toward an incoherent electronic state. Our results indicate that the suppression of the spin density wave order in CrBFA is unrelated to changes in the Fermi surface, and that the absence of superconductivity in both systems arises from competing magnetic effects: in CrBFA, the interplay between Cr local moments and Fe itinerant spin fluctuations, and in MnBFA, the collective magnetic impurity behavior of Mn spins. These findings highlight how disorder and local magnetic moments can hinder superconductivity in iron-based materials, even when nominal doping might suggest otherwise.
For further information:
- “Hole doping and electronic correlations in Cr substituted BaFe2Aa2”, Marli dos Reis Cantarino, Kevin R. Pakuszewski, Bjoern Salzmann, Pedro H. A. Moya, Wagner R. da Silva Neto, Gabriel S. Freitas, Pascoal G. Pagliuso, Cris Adriano, Walber H. Brito, Fernando A. Garcia, SciPost Phys. 17, 141 (2024).
- “Incoherent electronic band states in Mn-substituted BaFe2As2”, Marli R. Cantarino, Kevin R. Pakuszewski, Björn Salzmann, Pedro H. A. Moya, Wagner R. da Silva Neto, Gabriel S. Freitas, P. G. Pagliuso, Walber H. Brito, Claude Monney, C. Adriano, Fernando A. Garcia, Phys. Rev. B 108, 245124 (2023).
Connection between Defects and the Electronic Properties of Bi4I4:

Bismuth iodide (Bi4I4) is a quasi-one-dimensional material with topological and superconducting properties, whose electrical resistivity varies with its thermal history. Using a combined approach of transport measurements, performed by collaborators from University of São Paulo (USP), and density functional theory (DFT) calculations, we investigated the role of native point defects and mercury (Hg) impurities in the electronic properties of this material. Our results elucidate how the presence of these defects and impurities, such as antisites and vacancies, modifies the amount and type of charge carriers, directly impacting the resistivity profile of Bi4I4.
“Role of native point defects and Hg impurities in the electronic properties of Bi4I4”, Gustavo H. Cassemiro, C. David Hinostroza, Leandro Rodrigues de Faria, Daniel A. Mayoh, Jie Liu, Maria C. O. Aguiar, Martin R. Lees, Geetha Balakrishnan, J. Larrea Jiménez, Antonio Jefferson da Silva Machado, Valentina Martelli, Walber H. Brito, Phys. Rev. B 110, 224113 (2024).
Intrinsic topology in SrNbO3 thin films:
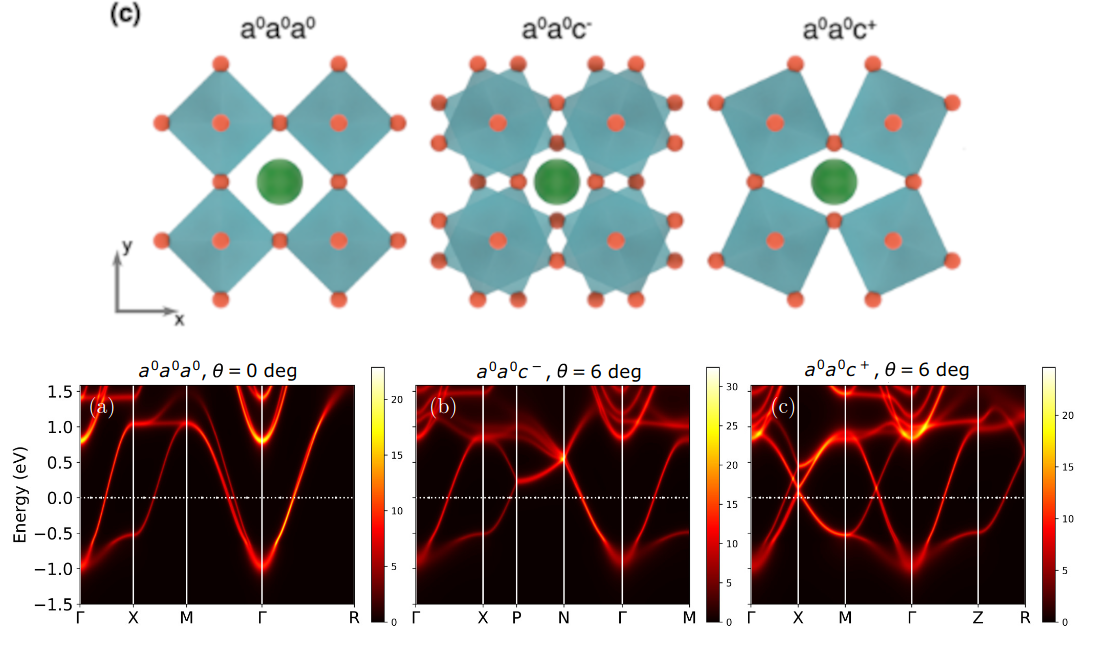
Strontium niobate (SrNbO₃) has emerged as a promising platform for transparent electronics and the realization of correlated Dirac semimetals. By combining density functional theory (DFT), dynamical mean-field theory (DMFT), and angle-resolved photoemission spectroscopy (ARPES), we reveal how octahedral tilting, controlled via strain, fundamentally shapes its electronic topology. Compressive strain favors out-of-plane tilting, while tensile strain induces in-plane rotations, with competing distorted phases hosting semi-Dirac dispersions that dynamical correlations shift toward the Fermi level. ARPES measurements, supported by diffraction data, confirm that intrinsic a⁰a⁰c⁺ rotations lead to topologically protected Dirac band crossings, giving rise to massless fermions. This interplay between structural distortions, band topology, and correlated electronic states offers a powerful route to tune thermoelectric, optical, and quantum properties in transition metal oxides, paving the way for next-generation electronic and photonic technologies.
For further information:
- “Intrinsic three-dimensional topology in SrNbO3 films”, A. Chikina, V. Rosendal, H. Li, E. Skoropata, E. B. Guedes, M. Caputo, N. C. Plumb, M. Shi, D. H. Petersen,M. Brandbyge, W. H. Brito, E. Pomjakushina, V. Scagnoli, J. Lyu, M. Medarde, U. Staub, S.-W. Huang, E. A. Müller Gubler, F. Baumberger, N. Pryds, and M. Radovic, Physical Review B 111, 155146 (2025).
- “Octahedral distortions in SrNbO3: Unraveling the structure-property relation”, Victor Rosendal, Walber H. Brito, Milan Radovic, Alla Chikina, Mads Brandbyge, Nini Pryds, Dirch H. Petersen, Phys. Rev. Materials 7, 075002 (2023).
Electronic Structure of correlated semiconductors (FeSb2):
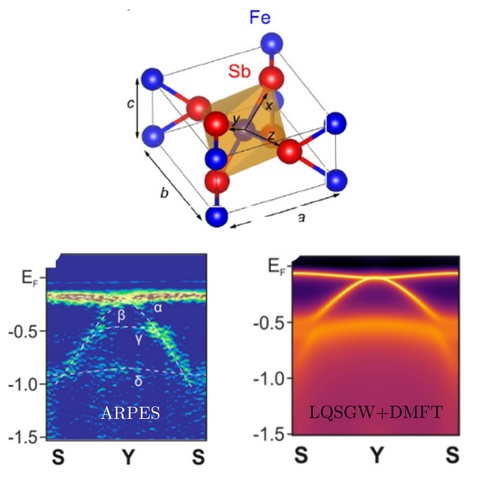
Some intermetallic compounds exhibit unusual semiconducting phases at low temperatures, which can even lead to large thermoelectric responses. For example, FeSb2 shows a colossal thermopower at low temperatures, along with a correlated semiconducting phase. Using GW+DMFT calculations, we investigate the electronic structure of FeSb2 in comparison with angle-resolved photoemission spectroscopy (ARPES) measurements, aiming to clarify the electronic states that give rise to its thermoelectric properties of interest.
For further information:
“Correlated electronic structure of colossal thermopower FeSb2: an ARPES and ab initio study”, A. Chikina, J.-Z. Ma, W.H. Brito, S. Choi, P. Sémon, A. Kutepov, Q. Du, J. Jandke, H. Liu, N.C. Plumb, M. Shi, C. Petrovic, M. Radovic, and G. Kotliar, Phys. Rev. Research 2, 023190 (2020).
Metal-insulator transitions in VO2 and NbO2:
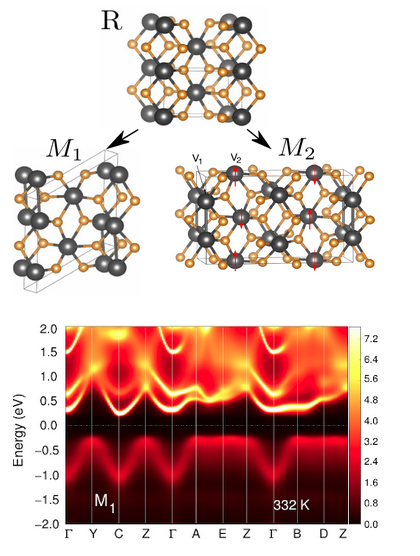
Vanadium (VO2) and niobium (NbO2) oxides exhibit metal–insulator transitions accompanied by significant structural changes. Using DFT combined with cluster DMFT calculations, we have elucidated how electronic correlations and structural distortions give rise to the insulating phases in these materials, as well as provided an accurate description of the correlated metallic phases observed at high temperatures.
For further information:
“Metal-Insulator Transition in VO2: a DFT+DMFT perspective“, W.H. Brito, M.C.O. Aguiar, K. Haule, and G. Kotliar, Phys. Rev. Lett. 117, 056402 (2016).
“Dynamic electronic correlation effects in NbO2 as compared to VO2”, W.H. Brito, M.C.O. Aguiar, K. Haule, and G. Kotliar, Phys. Rev. B 96, 195102 (2017).